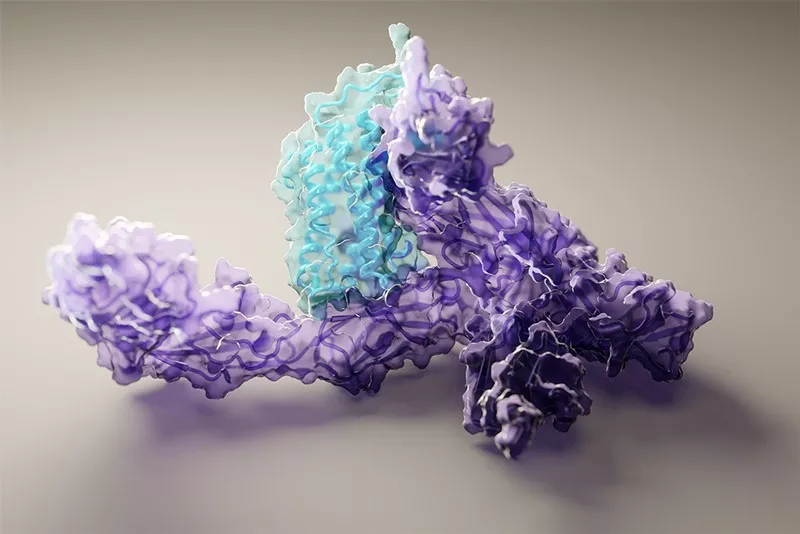
Компания DeepMind выпустила версию своей нейронной сети с глубоким обучением AlphaFold 2 для определения трехмерной структуры белков с открытым исходным кодом. Разработчики также подробно описали, как работает модель.
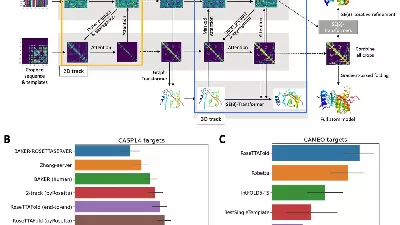
Тем временем академическая группа разработала собственный инструмент для определения формы белков, вдохновленный AlphaFold 2. RoseTTaFold определяет структуру белков почти так же хорошо, как AlphaFold 2. Его работу описали в статье в Science.
«Открытый исходный код инструментов означает, что научное сообщество имеет возможность использовать достижения для создания еще более мощного и полезного программного обеспечения», — говорит Дзинбо Сюй, вычислительный биолог из Чикагского университета в Иллинойсе.
Белки состоят из цепочек аминокислот, которые, будучи сложены в трехмерные формы, определяют функцию этих белков в клетках. На протяжении десятилетий исследователи использовали экспериментальные методы, такие как рентгеновская кристаллография и криоэлектронная микроскопия. Но такие методы могут быть трудоемкими и дорогостоящими, а некоторые белки не поддаются подобному анализу.
DeepMind в 2020 году показала, как ее программное обеспечение может точно предсказывать структуру многих белков, используя только их последовательность, которая определяется ДНК. Исследователи работали над своей системой в течение десятилетий, и AlphaFold 2 отлично показала себя в рамках критической оценки прогнозирования структуры белка CASP, решив 50-летнюю проблему фолдинга или «сворачивания» белков.
DeepMind кратко описала работу AlphaFold 2 в презентации на CASP 1 декабря. Компания пообещала опубликовать документы с более подробной информацией и сделать программное обеспечение доступным для исследователей.
Однако никаких подробностей о том, когда и как это произойдет, не последовало. В этот период Дэвид Бейкер, биохимик из Вашингтонского университета в Сиэтле, специалист по вычислительной химии Минкён Бэк и другие исследователи начали поиск способов повторить успех AlphaFold 2. Они определили, как сеть использует информацию о цепочках белков, и как предсказанные структуры одной части белка могут влиять на то, как сеть обрабатывает последовательности, соответствующие другим частям.
RoseTTaFold работает почти так же хорошо, как AlphaFold 2, и намного лучше, чем другие участники CASP. Как отмечает Бэк, в отличие от DeepMind, в лаборатории исследователей нет инженеров, занимающихся глубоким обучением. Именно этим объясняется то, что RoseTTaFold немного уступает AlphaFold 2.
Между тем команда Бейкера создала сервер, где исследователи могут разместить последовательность белка и получить предсказанную структуру. С момента запуска в прошлом месяце он уже предсказал структуру более 5 тысяч белков от 500 исследователей.
Хотя исходный код AlphaFold 2 находится в свободном доступе, в том числе для коммерческих организаций, он пока не может быть особенно полезным для исследователей без технических знаний. DeepMind сотрудничал с исследователями и организациями, в том числе с некоммерческой инициативой «Лекарства от забытых болезней», но теперь надеется расширить сотрудничество.
Источник: habr.com
Все права защищены.
Полное или частичное копирование материалов Сайта в коммерческих целях разрешено только с письменного разрешения владельца Сайта.
В случае обнаружения нарушений, виновные лица могут быть привлечены к ответственности в соответствии с действующим законодательством Российской Федерации.
